一个更好的检测PPCPs的方法,再生水将更安全
针对北京市部分农灌用再生水进行了分析,利用固相萃取(SPE)方法富集再生水中的污染物,UPLC-TOF-MS采集数据后,结合已有的污染物数据库进行筛查,对其种含量较高的色谱峰进行归属,确定了再生水中的16种含量较高的主要污染物,并结合UPLC-MS/MS对其进行定量分析。确定了一种有效的再生水中PPCPs筛查的方法。
1 实验部分
1.1药品与试剂
15种PPCPs标准品,包括阿奇霉素、甘宝素、氧氟沙星、美托洛尔、普罗帕酮、罗红霉素、替米沙坦、左西替利嗪、厄贝沙坦、莠去津、扑草净、烯酰吗啉、罗丹明B、格列齐特、三丁氧基乙基磷酸酯(纯度≥95%, Dr. Ehrenstorfer公司),三氯甲烷(CHCl3)(色谱纯,西陇化工股份有限公司),甲醇(MeOH)、乙腈(Ace)(色谱纯,Optima公司),甲酸(FA)(色谱纯,Alfa Aesar公司),超纯水。
1.2仪器与设备
HPLC Xevo G2-S QTOF超高效液相色谱-四极杆飞行时间质谱仪(美国Waters公司),配Electrospary Ion Source(ESI)源,Acquity UPLC BEH C18 色谱柱(2.1mm*100mm,1.7μm,美国Waters公司);N-EVAP112氮吹浓缩仪(美国Organomation公司),Vortex-Genie 2涡旋搅拌器(美国Scientific Industries公司),离心机,Milli-Q超纯水仪(美国Millipore公司),C18固相萃取柱(6cc,500mg,美国Waters公司)。
1.3样品前处理
1.3.1样品提取
将固相萃取柱依次用20mL三氯甲烷,20mL甲醇和10mL超纯水活化,1L样品水溶液以5mL/min的流速过柱,20mL超纯水淋洗,抽干,并用6mL甲醇洗脱目标物,收集洗脱液,于40℃下在氮吹浓缩装置上吹干。用甲醇-水(1:1,V:V)溶液定容至1mL,过0.22μm滤膜后待测。
1.3.2溶液配制
15种PPCPs标准品,包括。称取10mL标准品于10mL容量瓶中,以甲醇定容至刻度,将各标准品配制成1000mg/L标准储备液,4℃下保存。用甲醇-水(1:1,V:V)稀释,得1mg/L混合标准储备液。
1.4仪器参数
1.4.1液相色谱条件
BEH C18色谱柱(2.1mm*100mm,1.7μm);流动相:0.1%甲酸水溶液(A)和乙腈(B);流速:0.4mL/min;柱温:40℃;进样量:5μL;梯度洗脱程序:0~2min,3%B;2~14min,3%~100%B;14~15min,100%B;15~15.5min,100%~3%B;15.5~18min,3%B。
1.4.2质谱条件
全扫描(Full Scan)模式条件:正模式(ESI+)下采集数据,质谱参数设置为:全扫描范围:150-2000毛细管电压:3kV,源温:120℃,脱溶剂气流速:800L/h,脱溶剂气温度:450℃,低碰撞能量:5V,高碰撞能量范围:15-45V,实时质量校正溶液:亮氨酸-脑啡肽(0.4ng/μL,556.2766m/z)。数据采集与处理,碎片离子数据库建立均通过Waters UNIFI 1.7 software软件完成。
多反应监测(MRM)模式条件:正模式(ESI+)下采集数据,MRM参数见表1。数据采集与处理通过WatersMasslynx V4.1 software软件完成。
表1 16种PPCPs的MRM质谱参数
|
物质名称 |
分子式 |
母离子 (m/z) |
子离子 (m/z) |
碰撞能量 (eV) |
锥孔电压 (V) |
|
阿奇霉素 |
C38H72N2O12 |
749.8 |
158.2* |
43 |
15 |
|
591.6 |
40 |
15 |
|||
|
甘宝素 |
C15H17ClN2O2 |
293.2 |
68.9* |
19 |
15 |
|
197.1 |
25 |
15 |
|||
|
氧氟沙星 |
C18H20FN3O4 |
362.5 |
261.3* |
30 |
25 |
|
318.4 |
28 |
25 |
|||
|
美托洛尔 |
C15H25NO3 |
268.3 |
73.9* |
28 |
30 |
|
116 |
28 |
30 |
|||
|
自克威 |
C12H18N2O2 |
223.2 |
136.2* |
30 |
20 |
|
150.2 |
28 |
20 |
|||
|
普罗帕酮 |
C21H27NO3 |
342.3 |
98.2* |
20 |
20 |
|
116.2 |
24 |
20 |
|||
|
罗红霉素 |
C41H76N2O15 |
837.7 |
158.2* |
30 |
20 |
|
679.6 |
28 |
20 |
|||
|
替米沙坦 |
C33H30N4O2 |
515.5 |
276.3* |
48 |
10 |
|
497.4 |
45 |
10 |
|||
|
左西替利嗪 |
C21H25ClN2O3 |
389.5 |
166.2* |
38 |
18 |
|
201.2 |
17 |
18 |
|||
|
厄贝沙坦 |
C25H28N6O |
429.6 |
195.3* |
22 |
24 |
|
207.2 |
25 |
24 |
|||
|
莠去津 |
C8H14ClN5 |
216.4 |
96.2* |
25 |
25 |
|
174.2 |
20 |
25 |
|||
|
扑草净 |
C10H19N5S |
242.2 |
158.1* |
25 |
22 |
|
200.2 |
25 |
22 |
|||
|
烯酰吗啉 |
C21H22ClNO4 |
388.4 |
165.2* |
30 |
25 |
|
301.3 |
25 |
25 |
|||
|
罗丹明 B |
C28H31ClN2O3 |
443.5 |
399.5* |
50 |
35 |
|
385.4 |
50 |
35 |
|||
|
格列齐特 |
C15H21N3O3S |
324.5 |
127.1* |
23 |
25 |
|
153.2 |
25 |
25 |
|||
|
三丁氧基乙基磷酸酯 |
C18H39O7P |
399.3 |
99* |
22 |
12 |
|
143.1 |
20 |
12 |
|||
|
*:定量离子 |
|||||
2结果与讨论
2.1污染物的确证
2.1.1数据库筛查
在总离子流图中,利用一个包含345种污染物的数据库对样品中的污染物进行筛查,为了减少假阳性和假阴性的错误,对筛查参数进行了设置:色谱相应强度≥2000,质量误差≤5ppm,保留时间误差≤0.1min,高能量碎片匹配数量≥1,同位素匹配强度≤20%。在筛查参数的范围内,符合条件的污染物被确定并列出列表,为了进一步确认列表中的污染物,实验利用标准品质谱图与样品中的目标污染物的质谱图碎片进行对比。以罗红霉素为例(图1),经过设定条件的筛查,8.05min处837.6m/z的分子离子峰被归属为罗红霉素。我们以罗红霉素标准品进行验证,得到分子离子峰为837.6m/z,两个主要的碎片离子为679.6m/z和158.2m/z,和样品中的碎片离子一致,色谱图中,保留时间同为8.05min。
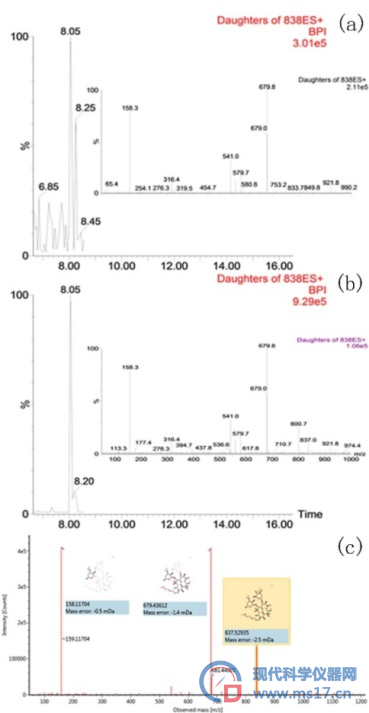
其中:(a)MRM条件下样品中罗红霉素的色谱图和质谱碎片;(b)MRM条件下罗红霉素标准品的色谱图和质谱碎片;(c)罗红霉素主要碎片的断裂方式推断。
图1 样品中罗红霉素的确认
2.1.2Chemspider搜索
对于响应强度很高但数据库中没有的污染物,实验利用Chemspider数据库进行检索。首先,我们根据精确质量数进行元素匹配,由于PPCPs类污染物通常含有C、H、O、N、P、S、F、Cl和Br等元素,我们首先利用“元素匹配”功能结合质谱中精确质量数对可能的元素组成进行分析,然后结合匹配结果中的分子式,利用Chemspider数据库进行搜索,筛选出可能存在的污染物。最后,高能量质谱图中的碎片离子被用来进一步确证疑似污染物。

其中:(a)UPLC-TOF-MS中全扫描模式下低能量色谱图;(b)UPLC-TOF-MS中全扫描模式下高能量色谱图;(c)色谱图中7.44min对应的低能量下的质谱碎片;(d)色谱图中7.44min对应的高能量下的质谱碎片;(e)替米沙坦主要碎片的断裂方式推断。
图2 样品中7.44min处未知物的推断
以7.44min的高响应值色谱峰为例(图2),在低能量质谱图中,有515.2m/z和305.2m/z两个主要的分子离子碎片,在高能量质谱图中,305.2m/z的响应强度几乎不变,只有515.2m/z的响应减弱,因此新增加的497.2m/z和276.1m/z可以被认为是由515.2m/z产生的碎片离子,我们选择515.2m/z作为分子离子峰,经过元素匹配得到可能的分子式为C33H30N4O2,然后通过Chemspider检索得到与之相符的替米沙坦的信息。最后,实验用替米沙坦标准品(515.2m/z)作对比,在相同条件下,替米沙坦标准品在7.44min得到色谱峰,高能量质谱图中产生497.2m/z和276.1m/z的碎片,进一步确证推断的准确性。
最后,经过对谱图的解析,16种主要的污染物被确定,16种污染物的具体信息见表2。
表 2 再生水中16种PPCPs的信息
|
物质名称 |
分子式 |
CAS号 |
分子量 (m/z) |
响应时间 (min) |
|
阿奇霉素 |
C38H72N2O12 |
83905-01-5 |
748.51 |
7.97 |
|
甘宝素 |
C15H17ClN2O2 |
38083-17-9 |
292.1 |
7.93 |
|
氧氟沙星 |
C18H20FN3O4 |
82419-36-1 |
361.14 |
5.08 |
|
美托洛尔 |
C15H25NO3 |
37350-58-6 |
267.18 |
5.7 |
|
自克威 |
C12H18N2O2 |
315-18-4 |
222.14 |
6.75 |
|
普罗帕酮 |
C21H27NO3 |
54063-53-5 |
341.2 |
7.79 |
|
罗红霉素 |
C41H76N2O15 |
80214-83-1 |
836.52 |
8.06 |
|
替米沙坦 |
C33H30N4O2 |
144701-48-4 |
514.24 |
7.44 |
|
左西替利嗪 |
C21H25ClN2O3 |
130018-77-8 |
388.16 |
7.94 |
|
厄贝沙坦 |
C25H28N6O |
138402-11-6 |
428.23 |
7.47 |
|
莠去津 |
C8H14ClN5 |
1912-24-9 |
215.09 |
8.05 |
|
扑草净 |
C10H19N5S |
7287-19-6 |
241.14 |
8.1 |
|
烯酰吗啉 |
C21H22ClNO4 |
110488-70-5 |
387.12 |
9.08 |
|
罗丹明B |
C28H31ClN2O3 |
81-88-9 |
478.2 |
9.37 |
|
格列齐特 |
C15H21N3O3S |
21187-98-4 |
323.13 |
8.98 |
|
三丁氧基乙基磷酸酯 |
C18H39O7P |
78-51-3 |
398.24 |
11.38 |
其中:(a)空白样品中,分别使用5mL、10mL、20mL活化试剂时,固相萃取柱中杂质的响应强度;(b)加标样品中,分别使用5mL、10mL、20mL活化试剂时,固相萃取柱中杂质的响应强度
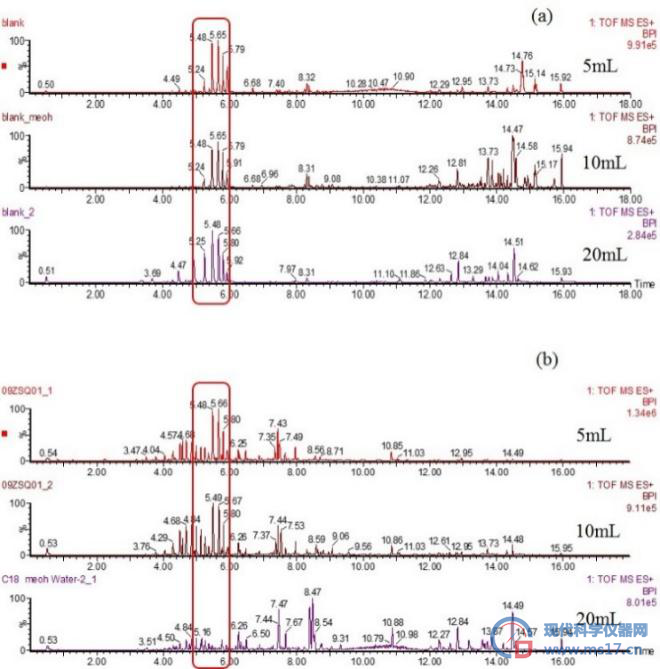
图3 不同体积活化试剂对固相萃取柱的杂质去除效果。
2.2前处理方法优化
再生水中污染物的低含量决定了对其进行富集的必要性,污染物的复杂多样决定了富集需要尽可能全面。为了对筛查出的PPCPs进行定量分析,实验通过加标方法对SPE前处理条件进行了优化。
1.阿奇霉素、2.甘宝素、3.氧氟沙星、4.美托洛尔、5.自克威、6.普罗帕酮、7.罗红霉素、8.替米沙坦、9.左西替利嗪、10.厄贝沙坦、11.莠去津、12.扑草净、13.烯酰吗啉、14罗丹明B、15.格列齐特、16.三丁氧基乙基磷酸酯
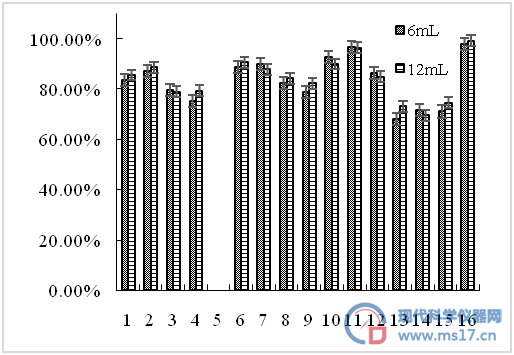
图4 洗脱液体积对PPCPs污染物的回收率影响
2.2.1活化液体积的优化
在固相萃取过程中,首先要去除固相萃取柱中的杂质成分,我们使用二氯甲烷和甲醇作为活化试剂分别除去固相萃取柱中的极性和非极性杂质,实验考察了分别用5mL、10mL、20mL作为活化试剂体积时,固相萃取柱中杂质的除去效果,结果见图3,当使用20mL二氯甲烷和20mL甲醇进行活化时,对固相萃取柱中的杂质出去效果较好。在空白样品中,5min~6min时固相萃取柱中的杂质响应值降低了3倍;在加标样品中,5min~6min时固相萃取柱中的杂质响应值降低程度大大减少了对目标污染物的干扰。
2.2.2洗脱液体积的优化
本实验采用甲醇作为固相萃取的洗脱溶剂。实验考察了不同洗脱液体积对目标污染物的净化程度,当洗脱液体积由6mL增大至12mL时,洗脱液中PPCPs的回收率几乎不变,最大提高程度约为5%(图4)。考虑到洗脱液体积的增大,后续使用氮气浓缩过程中的时间成本问题,实验选用6mL甲醇作为洗脱溶剂。
表3 16种PPCPs的标准曲线,线性相关系数,检出限,定量限,添加回收率范围
|
物质名称 |
标准曲线 |
线性相关系数 |
检出限 (ng/L) |
定量限 (ng/L) |
添加水平回收率 |
|||
|
5ng/L |
100ng/L |
500ng/L |
||||||
|
阿奇霉素 |
y=4.0655x+190.92 |
0.9903 |
10 |
8 |
/ |
83.48% |
78.78% |
|
|
甘宝素 |
y=23.6556x+49.2977 |
0.9923 |
0.8 |
0.5 |
89.68% |
87.35% |
90.93% |
|
|
氧氟沙星 |
y=11.1234x+19.9745 |
0.9919 |
10 |
8 |
/ |
79.52% |
82.47% |
|
|
美托洛尔 |
y=9.30674x-37.7541 |
0.9981 |
2.5 |
2 |
65.64% |
75.37% |
86.81% |
|
|
自克威 |
/ |
/ |
/ |
/ |
/ |
/ |
/ |
|
|
普罗帕酮 |
y=342.065x+156.809 |
0.9905 |
0.4 |
0.2 |
92.38% |
88.93% |
87.24% |
|
|
罗红霉素 |
y=44.407x+41492 |
0.9902 |
0.5 |
0.3 |
96.80% |
90.21% |
86.48% |
|
|
替米沙坦 |
y=4.97101x+32.5058 |
0.9909 |
0.8 |
0.5 |
79.92% |
82.47% |
89.61% |
|
|
左西替利嗪 |
y=67.2619x+68.1464 |
0.9936 |
0.8 |
0.5 |
80.29% |
79.06% |
83.42% |
|
|
厄贝沙坦 |
y=81.846x+47966 |
0.9928 |
0.5 |
0.3 |
98.05% |
92.81% |
108.88% |
|
|
莠去津 |
y=136.943x+171.712 |
0.9934 |
0.5 |
0.3 |
94.87% |
96.59% |
78.21% |
|
|
扑草净 |
y=176.644x+213.98 |
0.9906 |
0.8 |
0.5 |
84.37% |
86.48% |
84.56% |
|
|
烯酰吗啉 |
y=230.505x+74.9295 |
0.9983 |
0.8 |
0.5 |
71.89% |
68.03% |
66.94% |
|
|
罗丹明B |
y=554.166x+201.426 |
0.9991 |
0.3 |
0.2 |
86.69% |
71.53% |
84.91% |
|
|
格列齐特 |
y=47.7781x+218.02 |
0.9988 |
0.8 |
0.5 |
71.86% |
71.12% |
73.53% |
|
|
三丁氧基乙基磷酸酯 |
y=90.6965x+211.66 |
0.9965 |
0.5 |
0.3 |
81.15% |
97.91% |
92.39% |
|
|
n.d.指低于检出限,未被检测到. |
||||||||
2.3污染物的定量分析
16种污染物中,除自克威属于限制类药品,无法对其进行定性定量分析,实验通过UPLC-MS/MS的MRM模式结合Waters MassLynx软件对其余15种主要污染物进行了定量分析。为得到待测物质的最佳质谱条件,分别配置15种PPCPs的1mg/L标准溶液,直接进行质谱分析。一级质谱全扫描确定其分子离子峰,在二级质谱扫描条件下选择相对丰度较高的2个碎片离子作为定量和定性离子,分别优化各个离子对的锥孔电压和碰撞能量,其最优条件见表1。
通过方法学考察得到15种PPCPs的标准曲线,线性相关系数,检出限,定量限,添加回收率范围分别见表3。
2.4实际样品分析
实验采集了不同时期北京市24个不同地点的再生水样品,几乎所有物质在我们采集的水样品中均有检出,最大含量范围为6ng/L-4.9μg/L(表4)。
Table 4 The concentrations of 18 pollutants in wastewater samples
|
物质名称 |
浓度(ng/L) |
物质名称 |
浓度(ng/L) |
||||
|
最大值 |
最小值 |
平均值 |
最大值 |
最小值 |
平均值 |
||
|
阿奇霉素 |
112.79 |
n.d. |
37.366 |
左西替利嗪 |
132.28 |
1.986 |
30.016 |
|
甘宝素 |
347.29 |
n.d. |
107.644 |
厄贝沙坦 |
2041.6 |
n.d. |
393.654 |
|
氧氟沙星 |
221.52 |
n.d. |
26.351 |
莠去津 |
20517.4 |
1.191 |
3282.71 |
|
美托洛尔 |
888.73 |
3.885 |
118.117 |
扑草净 |
205.30 |
n.d. |
21.865 |
|
自克威 |
/ |
/ |
/ |
烯酰吗啉 |
104.40 |
1.035 |
28.569 |
|
普罗帕酮 |
6.012 |
n.d. |
1.632 |
罗丹明B |
83.950 |
0.314 |
19.487 |
|
罗红霉素 |
289.37 |
n.d. |
42.344 |
格列齐特 |
536.88 |
n.d. |
72.609 |
|
替米沙坦 |
4937.1 |
23.262 |
1455.68 |
三丁氧基乙基磷酸酯 |
183.72 |
2.573 |
42.957 |
3结论
本实验采用SPE-UPLC-TOF-MS方法对再生水中PPCPs污染物进行筛查。经过C18固相萃取柱富集,使用超高效液相色谱飞行时间质谱仪对样品中PPCPs类污染物进行筛查,并针对其中较高含量的15种污染物利用超高效液相色谱串联质谱进行定量分析。实验结果为污水处理和环境风险评估提供了有效可靠的方法。
(资讯来源《现代科学仪器》期刊,由“现代科学仪器网”官方发布,转载请注明来源)

关注本网官方微信 随时订阅权威资讯
